



Rotamers in Nitrosamines
ANÁLISE DE RISCO E IMPUREZAS
11/14/20257 min read
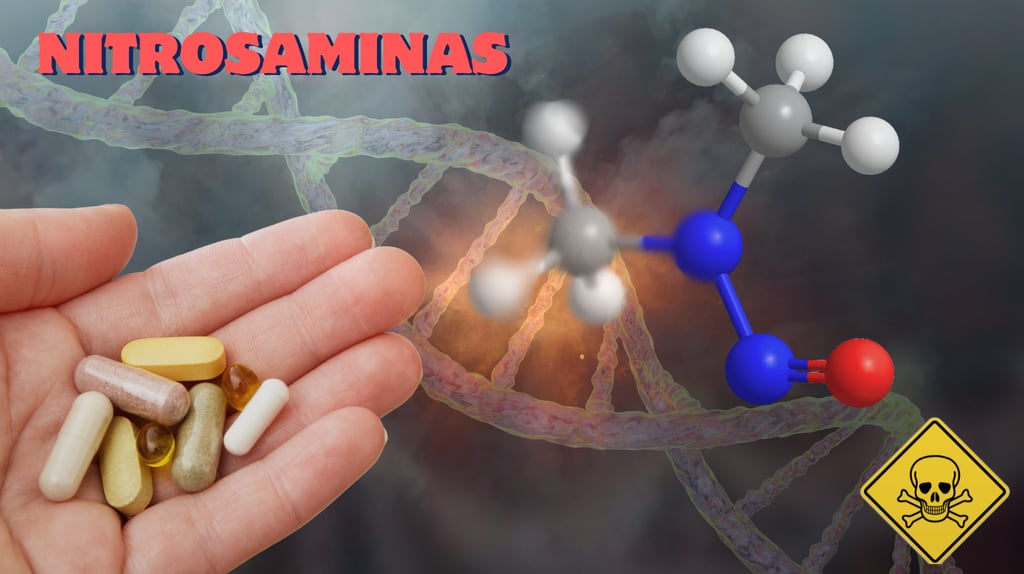

In the pharmaceutical industry, the detection and quantification of impurities such as nitrosamine drug substance-related impurities (NDSRI) is crucial to ensure the safety of medications.
A little-discussed phenomenon, but one that can significantly complicate analyses, is the presence of rotamers in nitrosamines. These conformers, resulting from molecular rotation, can lead to errors in chromatographic methods, affecting the accuracy of quantification.
In this post, we will explore in detail what rotamers are, how they form in nitrosamines, their interferences in analyses, and how to identify when a nitrosamine will exhibit this behavior. Based on scientific evidence, we offer practical insights for professionals in the field, aligned with the analytical consulting services of BizaLumière.
What Are Rotamers?
Rotamers, or rotational conformers, are conformational isomers that differ only by rotation around a single bond in a molecule. Unlike constitutional isomers or stereoisomers, rotamers do not involve changes in atomic connectivity, but rather in the spatial orientation of functional groups. They exist in dynamic equilibrium, with rotational energy barriers that determine their relative stability.
In organic chemistry, rotamers are common in molecules with C-C or N-N bonds, where rotation is restricted by steric or electronic interactions. For example, in compounds like butane, the gauche and anti rotamers are well-known. Thermal energy (kT) allows interconversion between these conformers, but under analytical conditions, such as in chromatography, they can be resolved as separate peaks if the rotational barrier is high enough.
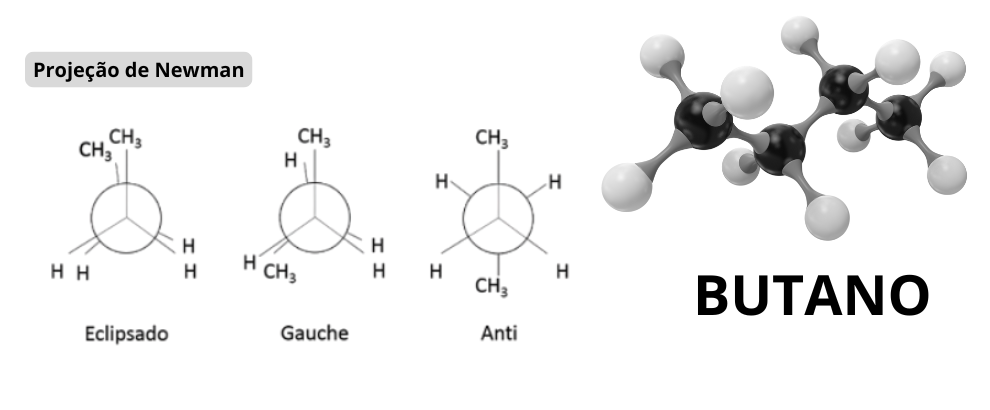
As described in "Conformational Analysis of Organic Compounds" (Eliel et al., 2010), rotamers are defined by rotational barriers typically between 1-10 kcal/mol, allowing rapid interconversion at room temperature, but potentially separable in analytical techniques.
Formation of Rotamers in Nitrosamines
Nitrosamines are compounds of the general form R1R2N-N=O, where the nitroso group (-N=N=O) is planar and linear. The N-N (nitro-amine) bond is a single bond, allowing rotation. In many nitrosamines, especially those with bulky alkyl groups (such as N-nitrosodimethylamine, NDMA, or N-nitrosodiisopropylamine, NDIPA), rotation around the N-N bond can generate distinct rotamers.
Mechanism of Formation
Molecular Structure: The nitroso group is planar due to resonance, but the N-N bond allows rotation. Groups R1 and R2 can be symmetric or asymmetric, influencing the rotational barrier. For example, in symmetric nitrosamines (R1 = R2), rotamers may be identical, but in asymmetric ones, they differ in energy.
Factors that Favor Formation:
Temperature: In chromatographic analyses, elevated temperatures (such as in GC-MS) can stabilize rotamers, while in LC-MS, polar solvents can influence the equilibrium.
Solvents: In organic or aqueous solutions, solvation can lower rotational barriers, allowing observation of multiple conformers.
Activation Energy: Typical barriers for rotation in nitrosamines are 5-15 kcal/mol, depending on substitution. Studies show that in NDMA, the barrier is low (~5 kcal/mol), but in cyclic nitrosamines, such as those in heterocycles, it can be higher.
In a study published in the Journal of the American Chemical Society (Vol. 142, 2020), researchers used NMR spectroscopy to demonstrate that nitrosamines like N-nitrosopiperidine exhibit rotamers due to N-N rotation, with barriers calculated via DFT (Density Functional Theory) around 10 kcal/mol.
Methods for Identification and Characterization of Rotamers
To distinguish and analyze rotamers in nitrosamines, it is necessary to employ precise and complementary methods. Advanced techniques allow obtaining data on molecular conformations, rotational dynamics, and chemical interactions, fundamental to understanding the properties and behavior of these molecules.
Spectroscopic Techniques
Nuclear Magnetic Resonance (NMR) is one of the most important tools for identifying rotamers, as it reveals differences in the chemical environment of atomic nuclei between conformations. The use of 1D and 2D spectra aids in mapping spatial interactions and distinguishing rotamers by their relaxation times and chemical shifts.
2D experiments such as NOESY and ROESY are particularly useful for detecting spatial proximities between nuclei, helping to differentiate rotamers that vary only in the relative orientation of functional groups.
Furthermore, variable-temperature NMR (VT-NMR) allows observing the interconversion between rotamers at different temperatures, enabling the calculation of rotational energy barriers (ΔG‡) and providing complementary thermodynamic and kinetic information.
In crystalline or semicrystalline samples, solid-state NMR can be used to distinguish rotamers in the solid state, revealing local interactions and molecular orientations that are not detectable in solution.
Infrared (IR) spectra also contribute to the identification of rotamers, detecting variations in bond vibrations caused by conformational changes. Meanwhile, mass spectrometry techniques (MS/MS) aid in structural confirmation, offering high sensitivity and specificity in the analysis of nitrosamines.
X-Ray Crystallography
X-ray crystallography provides precise three-dimensional structures of rotamers in the solid state, allowing visualization of the relative orientation of functional groups, bond angles, and other essential structural parameters. This method is especially useful when rotamers exhibit sufficient stability to form crystals.
The resolution obtained allows comparison of experimental data with theoretical and spectroscopic models, validating hypotheses about conformation and stability. Thus, crystallography serves as a solid structural basis for interpreting spectroscopic and computational results.
Computational Calculations
Theoretical simulations play a crucial role in the characterization of rotamers. Quantum methods, such as Density Functional Theory (DFT), allow predicting relative energies, rotational barriers, and optimized geometries of different conformers, enabling estimation of their stability and population.
Additionally, molecular dynamics (MD) techniques simulate the temporal evolution of conformations under real conditions of temperature and solvent, providing information on the frequency and relative stability of rotamers over time.
Computational models also help interpret experimental results, correlating spectra with structures, and provide detailed data on energy surfaces and intramolecular interactions relevant to the rotational dynamics of nitrosamines.
Factors Influencing the Formation of Rotamers in Nitrosamines
The formation of rotamers in nitrosamines directly depends on the molecular structure and external conditions that affect the stability and rotational freedom of chemical bonds. Factors such as spatial hindrances, chemical environment, and internal interactions play determining roles.
Steric Effects
The impact of bulky groups near bonds is decisive in restricting or allowing rotation of parts of the molecule. Larger groups create physical barriers that limit flexibility, resulting in distinct rotamers.
These steric effects influence both the speed and stability of rotational conformations. Molecules with bulky substituents near nitrogen tend to form more stable rotamers, as full rotation becomes energetically unfavorable.
In the context of nitrosamines, these conformational limitations can affect toxicity, chemical reactivity, and degradation mechanisms, since different rotamers may exhibit distinct behaviors.
Influence of the Medium
The chemical environment where the nitrosamine is found significantly affects the formation and stability of rotamers. Factors such as solvent type, pH, temperature, and the presence of chemical agents alter the conformational equilibrium.
In acidic media, for example, protonation or formation of the nitrosyl ion (NO⁺) modifies the electronic distribution, restricting or facilitating rotation around the N–N bond.
Higher temperatures increase available energy, allowing overcoming steric barriers and promoting interconversion between rotamers. Polar solvents with different polarities influence intermolecular interactions, altering spatial orientation and relative stability of conformations.
Intramolecular Interactions
Within the molecule, internal hydrogen bonds, dipoles, and Van der Waals forces can stabilize certain conformations and prevent free rotation. The presence of highly electronegative atoms nearby generates dipoles that attract or repel specific regions of the molecule, fixing one rotamer over another.
In nitrosamines, these intramolecular interactions control the electronic distribution and influence the relative energy of rotamers, affecting critical properties such as reactivity, thermal stability, and compound lifetime.
The detailed characterization of rotamers in nitrosamines is essential to understand their chemical and biological properties, including reactivity, stability, and toxicological potential.
The integration of spectroscopic techniques (NMR, IR, MS), structural methods (crystallography and solid-state NMR), and computational approaches (DFT, molecular dynamics) provides a comprehensive and accurate view of the structure, dynamics, and conformational behavior of these complex molecules.
Interferences in Nitrosamine Quantification Analyses
Rotamers can cause serious interferences in analytical methods used to quantify nitrosamines, such as gas chromatography coupled with mass spectrometry (GC-MS) and high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS). These methods are essential for detecting NDSRI in medications, in accordance with FDA, EMA, and Anvisa guidelines.
How They Interfere
Peak Separation: In chromatography, rotamers with sufficient rotational barriers (>10 kcal/mol) can elute as separate peaks, leading to underestimation or overestimation of concentration. For example, if a nitrosamine presents two rotamers in 1:1 proportions, the expected single peak may split, resulting in incorrect quantification.
Sensitivity and Specificity: In GC-MS, elevated injection temperatures can promote interconversion, but in LC-MS, solvents can stabilize conformers, causing duplicated peaks. This affects the limit of detection (LOD) and quantification (LOQ), which must be below 30 ng/day for nitrosamines, in accordance with EMA's Acceptable Intake (AI).
Practical Examples: In NDMA analyses in proton pump inhibitors, rotamers were observed in GC-MS, leading to errors of up to 20% in quantification if not corrected (Source: FDA Guidance on Nitrosamine Impurities, 2021). In LC-MS/MS, solvents like acetonitrile can induce rotamers in heterocyclic nitrosamines, complicating peak integration. Furthermore, in LC/MS-MS, fragmentation in MS-MS can be influenced by conformations, altering ion patterns or intensities, which is relevant for identifying nitrosamines in complex samples (e.g., environmental, pharmaceutical). Toxicological studies of nitrosamines (such as NDMA) often use LC/MS-MS for detection at low concentrations, and knowledge of rotamers helps interpret stability or reaction data.
An article in Analytical Chemistry (Vol. 93, 2021) discusses how rotamers in nitrosamines interfere in GC-MS, recommending isothermal conditions to minimize separation. Validation studies show that without correction, the relative error can exceed 15%.
How to Know When a Nitrosamine Will Have Rotamers
Identifying in advance whether a nitrosamine will exhibit rotamers is key to optimizing analyses. This depends on the molecular structure and can be predicted by theoretical calculations or observed experimentally.
Criteria for Identification
Chemical Structure: Nitrosamines with asymmetric alkyl groups (e.g., N-ethyl-N-methylnitrosamine) or heterocyclic ones (e.g., N-nitrosopyrrolidine) have a higher probability of rotamers due to steric interactions. In contrast, symmetric nitrosamines like NDMA rarely exhibit this phenomenon.
Theoretical Calculations: Use DFT to calculate rotational barriers. If >8 kcal/mol, rotamers are likely. Software like Gaussian or ORCA is useful for molecular modeling.
Spectroscopy: NMR (Nuclear Magnetic Resonance) is the standard technique for detecting rotamers, showing duplicated signals for equivalent protons. In MS, different fragmentation may indicate conformers.
Preliminary Chromatographic Analysis: Tests at different temperatures or solvents reveal multiple peaks. If the barrier is low, use derivatization to stabilize one conformer.
According to the ICH Q3A Guideline (International Council for Harmonisation), structural evaluations must include conformer predictions. A study in the Journal of Chromatography A (Vol. 1621, 2020) uses DFT to predict rotamers in nitrosamines, correlating with experimental data.
Implications and Control Strategies
Rotamers not only complicate analyses but also impact regulatory compliance, potentially leading to recalls if quantifications are inaccurate.
Strategies include:
Analytical Optimization: Use controlled temperatures or solvents that promote rapid interconversion. In GC-MS, gentle temperature ramps help.
Data Correction: Apply correction factors based on rotamer proportions or use total integration.
Method Validation: Include robustness tests for rotamers, in accordance with USP <1225> (Validation of Analytical Procedures).
EMA guidelines (2020) emphasize the need for methods robust against conformational interferences.
Conclusion
Rotamers in nitrosamines represent a technical challenge in pharmaceutical analyses, but with in-depth knowledge, they can be managed. Understanding their formation, interferences, and identification is essential to ensure accurate quantifications and compliance with global regulations.
If your company faces difficulties with NDSRI or similar analyses, BizaLumière is ready to help with personalized analytical consulting, utilizing solid knowledge in Nitrosamines.
Contact us for a free assessment and illuminate your data with expertise!
📩 contato@bizalumiere.com.br
🌐 www.bizalumiere.com/en
References
Eliel, E. L., et al. (2010). Conformational Analysis of Organic Compounds. Wiley.
FDA Guidance for Industry: Control of Nitrosamine Impurities in Human Drugs (2021).
EMA Guideline on Nitrosamine Impurities (2020).
Scientific articles: J. Am. Chem. Soc. (2020), Anal. Chem. (2021), J. Chromatogr. A (2020).
BizaLumière
Specialized consulting in Technical Documentation and Analytical Regulatory Requirements
ContaCT
newsletter
© 2025. All rights reserved.
BizaLumière - CNPJ: 61.773.029/0001-09
Mogi das Cruzes - SP
Privacy Policy